Benchmarking accuracy and precision of intensity-based absolute quantification of protein abundances in Saccharomyces cerevisiae
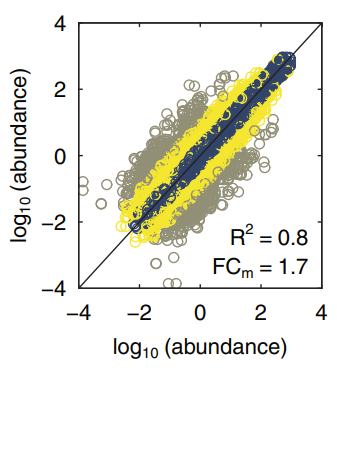 Variability across different batch runs of the mass spectrometer. Taken from the original publication: https://doi.org/10.1002/pmic.202000093
Variability across different batch runs of the mass spectrometer. Taken from the original publication: https://doi.org/10.1002/pmic.202000093Abstract
This was a side project in my PhD, as I needed to measure the exact amount of protein copy number inside a cell for combining it with my simulations, so I generated a dataset using label-free mass spectrometry proteomics. However, I saw that the technique had really poor technical reproducibility, i.e. it did not deliver consistent results when the same sample would be measured repeatedly. Therefore, we decided to perform a dedicated study of accuracy and precision of this technique by measuring proteomics data for yeast with both biological and inter-batch technical triplicates. We also analyzed how do the results vary when applying different methods for converting the raw data. Surprisingly, we demonstrated that a simple normalization and rescaling can perform as accurately, yet more precisely, than ‘state of the art’ methods that rely on expensive external standards, meaning that for achieving best results, it’s better not to use any external standard, and apply the suggested conversion instead. Additionally, we showed that inter-batch reproducibility is worse than biological reproducibility regardless of the method used, which is evidence of the current limitations of this technique. Even though the findings were quite controversial, I’m very happy we published this story, and even more so that we did it in a fully reproducible way, with all supplementary material being a rendered R markdown file.